

地址:广州开发区科研路2号4栋5楼
产品咨询:13392693259
耗材电话:18998328368
售后服务:020-87684117
传 真:020-87684846
Email:info@gdana.com
网址:http://www.gdana.com/
前言
本标准代替GB5009.12—2010《食品安全国家标准 食品中铅的测定》、GB/T20380.3—2006《淀粉及其制品 重金属含量 第3部分:电热原子吸收光谱法测定铅含量》、GB/T23870—2009《蜂胶中铅的测定 微波消解-石墨炉原 子吸收分光光度法》、GB/T18932.12—2002《蜂蜜中钾、钠、钙、镁、锌、铁、铜、锰、铬、铅、镉含量的测定方法 原子吸收光谱法》、NY/T1100—2006《稻米中铅、镉的测定 石墨炉原子吸收光谱法》,SN/T2211—2008《蜂皇浆中铅和镉的测定石墨炉原子吸收光谱法》中铅的测定方法。
本标准与GB5009.12—2010相比,主要变化如下:
——在前处理方法中,保留湿法消解和压力罐消解,删除干法灰化和过硫酸铵灰化法,增加微波消解;
——保留石墨炉原子吸收光谱法为第一法,采用磷酸二氢铵-硝酸钯溶液作为基体改进剂;保留火焰原子吸收光 谱法为第三法;保留二硫腙比色法为第四法;
——增加电感耦合等离子体质谱法作为第二法; ——删除氢化物原子荧光光谱法、单扫描极谱法;
——增加了微波消解升温程序、石墨炉原子吸收光谱法和火焰原子吸收光谱法的仪器参考条件为附录。
1、范围
本标准规定了食品中铅含量测定的石墨炉原子吸收光谱法、电感耦合等离子体质谱法、火焰原子吸收光谱法和二硫腙比色法。
本标准适用于各类食品中铅含量的测定。
第一法石墨炉原子吸收光谱法
2、原理
试样消解处理后,经石墨炉原子化,在283.3nm处测定吸光度。在一定浓度范围内铅的吸光度值与铅含量成正比,与标准系列比较定量。
3、试剂和材料
除非另有说明,本方法所用试剂均为优级纯,水为GB/T6682规定的二级水。
3.1试剂
3.1.1硝酸(HNO3)。
3.1.2高氯酸(HClO4)。
3.1.3磷酸二氢铵(NH4H2PO4)。
3.1.4硝酸钯[Pd(NO3)2]。
3.2试剂配制
3.2.1硝酸溶液(5+95):量取50mL硝酸,缓慢加入到950mL水中,混匀。
3.2.2硝酸溶液(1+9):量取50mL硝酸,缓慢加入到450mL水中,混匀。
3.2.3磷酸二氢铵-硝酸钯溶液:称取0.02g硝酸钯,加少量硝酸溶液(1+9)溶解后,再加入2g磷酸二氢铵,溶解后用硝酸溶液(5+95)定容至100mL,混匀。
3.3标准品 硝酸铅[Pb(NO3)2,CAS号:10099-74-8]:纯度>99.99%。或经国家认证并授予标准物质证书的一定浓度的铅标准溶液。
3.4标准溶液配制
3.4.1铅标准储备液(1000mg/L):准确称取1.5985g(精确至0.0001g)硝酸铅,用少量硝酸溶液(1+9)溶解,移入1000mL容量瓶,加水至刻度,混匀。 3.4.2铅标准中间液(1.00mg/L):准确吸取铅标准储备液(1000mg/L)1.00mL于1000mL容量瓶中,加硝酸溶液(5+95)至刻度,混匀。
3.4.3铅标准系列溶液:分别吸取铅标准中间液(1.00mg/L)0mL、0.500mL、1.00mL、2.00mL、3.00mL和4.00mL于100mL容量瓶中,加硝酸溶液(5+95)至刻度,混匀。此铅标准系列溶液的质量浓度分别为0μg/L、5.00μg/L、10.0μg/L、20.0μg/L、30.0μg/L和40.0μg/L。
注:可根据仪器的灵敏度及样品中铅的实际含量确定标准系列溶液中铅的质量浓度。
4、仪器和设备
注:所有玻璃器皿及聚四氟乙烯消解內罐均需硝酸溶液(1+5)浸泡过夜,用自来水反复冲洗,最后用水冲洗干净。
4.1原子吸收光谱仪:配石墨炉原子化器,附铅空心阴极灯。
4.2分析天平:感量0.1mg和1mg。
4.3可调式电热炉。
4.4可调式电热板。
4.5微波消解系统:配聚四氟乙烯消解内罐。
4.6恒温干燥箱。
4.7压力消解罐:配聚四氟乙烯消解内罐。
5、分析步骤
5.1试样制备
注:在采样和试样制备过程中,应避免试样污染。
5.1.1粮食、豆类样品
样品去除杂物后,粉碎,储于塑料瓶中。
5.1.2蔬菜、水果、鱼类、肉类等样品
样品用水洗净,晾干,取可食部分,制成匀浆,储于塑料瓶中。
5.1.3饮料、酒、醋、酱油、食用植物油、液态乳等液体样品
将样品摇匀。
5.2试样前处理
5.2.1湿法消解
称取固体试样0.2g~3g(精确至0.001g)或准确移取液体试样0.500mL~5.00mL于带刻度消化管中,加入10mL硝酸和0.5mL高氯酸,在可调式电热炉上消解(参考条件:120℃/0.5h~1h;升至180℃/2h~4h、升至200℃~220℃)。若消化液呈棕褐色,再加少量硝酸,消解至冒白烟,消化液呈无色透明或略带黄色,取出消化管,冷却后用水定容至10mL,混匀备用。同时做试剂空白试验。亦可采用锥形瓶,于可调式电热板上,按上述操作方法进行湿法消解。
5.2.2微波消解
称取固体试样0.2g~0.8g(精确至0.001g)或准确移取液体试样0.500mL~3.00mL于微波消解罐中,加入5mL硝酸,按照微波消解的操作步骤消解试样,消解条件参考附录A。冷却后取出消解罐,在电热板上于140℃~160℃赶酸至1mL左右。消解罐放冷后,将消化液转移至10mL容量瓶中,用少量水洗涤消解罐2次~3次,合并洗涤液于容量瓶中并用水定容至刻度,混匀备用。同时做试剂空白试验。
5.2.3压力罐消解
称取固体试样0.2g~1g(精确至0.001g)或准确移取液体试样0.500mL~5.00mL于消解内罐中,加入5mL硝酸。盖好内盖,旋紧不锈钢外套,放入恒温干燥箱,于140℃~160℃下保持4h~5h。冷却后缓慢旋松外罐,取出消解内罐,放在可调式电热板上于140℃~160℃赶酸至1mL左右。冷却后将消化液转移至10mL容量瓶中,用少量水洗涤内罐和内盖2次~3次,合并洗涤液于容量瓶中并用水定容至刻度,混匀备用。同时做试剂空白试验。
5.3测定
5.3.1仪器参考条件
根据各自仪器性能调至最佳状态。参考条件见附录B。
5.3.2标准曲线的制作
按质量浓度由低到高的顺序分别将10μL铅标准系列溶液和5μL磷酸二氢铵-硝酸钯溶液(可根据所使用的仪器确定最佳进样量)同时注入石墨炉,原子化后测其吸光度值,以质量浓度为横坐标,吸光度值为纵坐标,制作标准曲线。
5.3.3试样溶液的测定
在与测定标准溶液相同的实验条件下,将10μL空白溶液或试样溶液与5μL磷酸二氢铵-硝酸钯溶液(可根据所使用的仪器确定最佳进样量)同时注入石墨炉,原子化后测其吸光度值,与标准系列比较定量。
6、分析结果的表述
试样中铅的含量按式(1)计算:
X=(ρ-ρ0)×V/m×1000…………………………(1)
式中:
X——试样中铅的含量,单位为毫克每千克或毫克每升(mg/kg或mg/L);
ρ——试样溶液中铅的质量浓度,单位为微克每升(μg/L);
ρ0——空白溶液中铅的质量浓度,单位为微克每升(μg/L);
V——试样消化液的定容体积,单位为毫升(mL);
m——试样称样量或移取体积,单位为克或毫升(g或mL);
1000——换算系数。
当铅含量≥1.00mg/kg(或mg/L)时,计算结果保留三位有效数字;当铅含量<1.00mg/kg(或mg/L)时,计算结果保留两位有效数字。
7、精密度
在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%。
8、其他
当称样量为0.5g(或0.5mL),定容体积为10mL时,方法的检出限为0.02mg/kg(或0.02mg/L),定量限为0.04mg/kg(或0.04mg/L)。
第二法电感耦合等离子体质谱法
见GB5009.268。
第三法火焰原子吸收光谱法
9、原理
试样经处理后,铅离子在一定pH条件下与二乙基二硫代氨基甲酸钠(DDTC)形成络合物,经4-甲基-2-戊酮(MIBK)萃取分离,导入原子吸收光谱仪中,经火焰原子化,在283.3nm处测定的吸光度。在一定浓度范围内铅的吸光度值与铅含量成正比,与标准系列比较定量。
10、试剂和材料
注:除非另有说明,本方法所用试剂均为分析纯,水为GB/T6682规定的二级水。
10.1试剂
10.1.1硝酸(HNO3):优级纯。
10.1.2高氯酸(HClO4):优级纯。
10.1.3硫酸铵[(NH4)2SO4]。
10.1.4柠檬酸铵[C6H5O7(NH4)3]。
10.1.5溴百里酚蓝(C27H28O5SBr2)。
10.1.6二乙基二硫代氨基甲酸钠[DDTC,(C2H5)2NCSSNa·3H2O]。
10.1.7氨水(NH3·H2O):优级纯。
10.1.84-甲基-2-戊酮(MIBK,C6H12O)。
10.1.9盐酸(HCl):优级纯。
10.2试剂配制
10.2.1硝酸溶液(5+95):量取50mL硝酸,加入到950mL水中,混匀。
10.2.2硝酸溶液(1+9):量取50mL硝酸,加入到450mL水中,混匀。
10.2.3硫酸铵溶液(300g/L):称取30g硫酸铵,用水溶解并稀释至100mL,混匀。
10.2.4柠檬酸铵溶液(250g/L):称取25g柠檬酸铵,用水溶解并稀释至100mL,混匀。
10.2.5溴百里酚蓝水溶液(1g/L):称取0.1g溴百里酚蓝,用水溶解并稀释至100mL,混匀。
10.2.6DDTC溶液(50g/L):称取5gDDTC,用水溶解并稀释至100mL,混匀。 10.2.7氨水溶液(1+1):吸取100mL氨水,加入100mL水,混匀。
10.2.8盐酸溶液(1+11):吸取10mL盐酸,加入110mL水,混匀。
10.3标准品
硝酸铅[Pb(NO3)2,CAS号:10099-74-8]:纯度>99.99%。或经国家认证并授予标准物质证书的一定浓度的铅标准溶液。
10.4标准溶液配制
10.4.1铅标准储备液(1000mg/L):准确称取1.5985g(精确至0.0001g)硝酸铅,用少量硝酸溶液(1+9)溶解,移入1000mL容量瓶,加水至刻度,混匀。
10.4.2铅标准使用液(10.0mg/L):准确吸取铅标准储备液(1000mg/L)1.00mL于100mL容量瓶中,加硝酸溶液(5+95)至刻度,混匀。
11、仪器和设备
注:所有玻璃器皿均需硝酸(1+5)浸泡过夜,用自来水反复冲洗,最后用水冲洗干净。
11.1原子吸收光谱仪:配火焰原子化器,附铅空心阴极灯。
11.2分析天平:感量0.1mg和1mg。
11.3可调式电热炉。
11.4可调式电热板。
12、分析步骤
12.1试样制备
同5.1。
12.2试样前处理
同5.2.1
12.3测定
12.3.1仪器参考条件
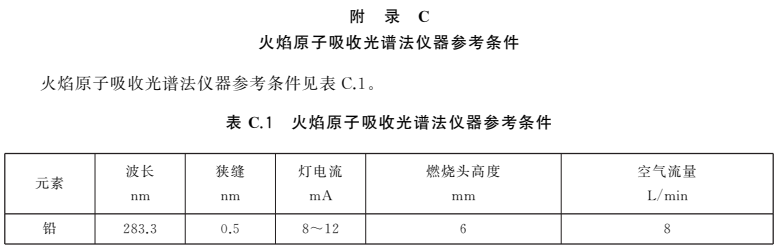
根据各自仪器性能调至最佳状态。参考条件参见附录C。
12.3.2标准曲线的制作
分别吸取铅标准使用液0mL、0.250mL、0.500mL、1.00mL、1.50mL和2.00mL(相当0μg、2.50μg、5.00μg、10.0μg、15.0μg和20.0μg铅)于125mL分液漏斗中,补加水至60mL。加2mL柠檬酸铵溶液(250g/L),溴百里酚蓝水溶液(1g/L)3滴~5滴,用氨水溶液(1+1)调pH至溶液由黄变蓝,加硫酸铵溶液(300g/L)10mL,DDTC溶液(1g/L)10mL,摇匀。放置5min左右,加入10mLMIBK,剧烈振摇提取1min,静置分层后,弃去水层,将MIBK层放入10mL带塞刻度管中,得到标准系列溶液。
将标准系列溶液按质量由低到高的顺序分别导入火焰原子化器,原子化后测其吸光度值,以铅的质量为横坐标,吸光度值为纵坐标,制作标准曲线。
12.3.3试样溶液的测定
将试样消化液及试剂空白溶液分别置于125mL分液漏斗中,补加水至60mL。加2mL柠檬酸铵溶液(250g/L),溴百里酚蓝水溶液(1g/L)3滴~5滴,用氨水溶液(1+1)调pH至溶液由黄变蓝,加硫酸铵溶液(300g/L)10mL,DDTC溶液(1g/L)10mL,摇匀。放置5min左右,加入10mLMIBK,剧烈振摇提取1min,静置分层后,弃去水层,将MIBK层放入10mL带塞刻度管中,得到试样溶液和空白溶液。
将试样溶液和空白溶液分别导入火焰原子化器,原子化后测其吸光度值,与标准系列比较定量。
13、分析结果的表述
试样中铅的含量按式(2)计算:
X=(m1-m0)/m2…………………………(2)
式中:
X———试样中铅的含量,单位为毫克每千克或毫克每升(mg/kg或mg/L);
m1———试样溶液中铅的质量,单位为微克(μg);
m0———空白溶液中铅的质量,单位为微克(μg);
m2———试样称样量或移取体积,单位为克或毫升(g或mL)。
当铅含量≥10.0mg/kg(或mg/L)时,计算结果保留三位有效数字;当铅含量<10.0mg/kg(或mg/L)时,计算结果保留两位有效数字。
14、精密度
在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%。
15、其他
以称样量0.5g(或0.5mL)计算,方法的检出限为0.4mg/kg(或0.4mg/L),定量限为1.2mg/kg(或1.2mg/L)。
第四法二硫腙比色法
16、原理
试样经消化后,在pH8.5~9.0时,铅离子与二硫腙生成红色络合物,溶于三氯甲烷。加入柠檬酸铵、氰化钾和盐酸羟胺等,防止铁、铜、锌等离子干扰。于波长510nm处测定吸光度,与标准系列比较定量。
17、试剂和材料
除非另有说明,本方法所用试剂均为分析纯,水为GB/T6682规定的三级水。
17.1试剂
17.1.1硝酸(HNO3):优级纯。
17.1.2高氯酸(HClO4):优级纯。
17.1.3氨水(NH3·H2O):优级纯。
17.1.4盐酸(HCl):优级纯。
17.1.5酚红(C19H14O5S)。
17.1.6盐酸羟胺(NH2OH·HCl)。
17.1.7柠檬酸铵[C6H5O7(NH4)3]。
17.1.8氰化钾(KCN)。
17.1.9三氯甲烷(CH3Cl,不应含氧化物)。
17.1.10二硫腙(C6H5NHNHCSN=NC6H5)。
17.1.11乙醇(C2H5OH):优级纯。
17.2试剂配制
17.2.1硝酸溶液(5+95):量取50mL硝酸,缓慢加入到950mL水中,混匀。
17.2.2硝酸溶液(1+9):量取50mL硝酸,缓慢加入到450mL水中,混匀。
17.2.3氨水溶液(1+1):量取100mL氨水,加入100mL水,混匀。
17.2.4氨水溶液(1+99):量取10mL氨水,加入990mL水,混匀。
17.2.5盐酸溶液(1+1):量取100mL盐酸,加入100mL水,混匀。
17.2.6酚红指示液(1g/L):称取0.1g酚红,用少量多次乙醇溶解后移入100mL容量瓶中并定容至刻度,混匀。
17.2.7二硫腙-三氯甲烷溶液(0.5g/L):称取0.5g二硫腙,用三氯甲烷溶解,并定容至1000mL,混匀,保存于0℃~5℃下,必要时用下述方法纯化。 称取0.5g研细的二硫腙,溶于50mL三氯甲烷中,如不全溶,可用滤纸过滤于250mL分液漏斗中,用氨水溶液(1+99)提取三次,每次100mL,将提取液用棉花过滤至500mL分液漏斗中,用盐酸溶液(1+1)调至酸性,将沉淀出的二硫腙用三氯甲烷提取2次~3次,每次20mL,合并三氯甲烷层,用等量水洗涤两次,弃去洗涤液,在50℃水浴上蒸去三氯甲烷。精制的二硫腙置硫酸干燥器中,干燥备用。或将沉淀出的二硫腙用200mL、200mL、100mL三氯甲烷提取三次,合并三氯甲烷层为二硫腙-三氯甲烷溶液。
17.2.8盐酸羟胺溶液(200g/L):称20g盐酸羟胺,加水溶解至50mL,加2滴酚红指示液(1g/L),加氨水溶液(1+1),调pH至8.5~9.0(由黄变红,再多加2滴),用二硫腙-三氯甲烷溶液(0.5g/L)提取至三氯甲烷层绿色不变为止,再用三氯甲烷洗二次,弃去三氯甲烷层,水层加盐酸溶液(1+1)至呈酸性,加水至100mL,混匀。
17.2.9柠檬酸铵溶液(200g/L):称取50g柠檬酸铵,溶于100mL水中,加2滴酚红指示液(1g/L),加氨水溶液(1+1),调pH至8.5~9.0,用二硫腙-三氯甲烷溶液(0.5g/L)提取数次,每次10mL~20mL,至三氯甲烷层绿色不变为止,弃去三氯甲烷层,再用三氯甲烷洗二次,每次5mL,弃去三氯甲烷层,加水稀释至250mL,混匀。
17.2.10氰化钾溶液(100g/L):称取10g氰化钾,用水溶解后稀释至100mL,混匀。
17.2.11二硫腙使用液:吸取1.0mL二硫腙-三氯甲烷溶液(0.5g/L),加三氯甲烷至10mL,混匀。用1cm比色杯,以三氯甲烷调节零点,于波长510nm处测吸光度(A),用式(3)算出配制100mL二硫腙使用液(70%透光率)所需二硫腙-三氯甲烷溶液(0.5g/L)的毫升数(V)。量取计算所得体积的二硫腙三氯甲烷溶液,用三氯甲烷稀释至100mL。
V=10×(2-lg70)/A=1.55/A…………………………(3)
17.3标准品
硝酸铅[Pb(NO3)2,CAS号:10099-74-8]:纯度>99.99%。或经国家认证并授予标准物质证书的一定浓度的铅标准溶液。
17.4标准溶液配制
同10.4。
18、仪器和设备
注:所有玻璃器皿均需硝酸(1+5)浸泡过夜,用自来水反复冲洗,最后用水冲洗干净。
18.1分光光度计。
18.2分析天平:感量0.1mg和1mg。
18.3可调式电热炉。
18.4可调式电热板。
19、分析步骤
19.1试样制备
同5.1。
19.2试样前处理
同5.2.1。
19.3测定
19.3.1仪器参考条件
根据各自仪器性能调至最佳状态。测定波长:510nm。
19.3.2标准曲线的制作
吸取0mL、0.100mL、0.200mL、0.300mL、0.400mL和0.500mL铅标准使用液(相当0μg、1.00μg、2.00μg、3.00μg、4.00μg和5.00μg铅)分别置于125mL分液漏斗中,各加硝酸溶液(5+95)至20mL。再各加2mL柠檬酸铵溶液(200g/L),1mL盐酸羟胺溶液(200g/L)和2滴酚红指示液(1g/L),用氨水溶液(1+1)调至红色,再各加2mL氰化钾溶液(100g/L),混匀。各加5mL二硫腙使用液,剧烈振摇1min,静置分层后,三氯甲烷层经脱脂棉滤入1cm比色杯中,以三氯甲烷调节零点于波长510nm处测吸光度,以铅的质量为横坐标,吸光度值为纵坐标,制作标准曲线。
19.3.3试样溶液的测定
将试样溶液及空白溶液分别置于125mL分液漏斗中,各加硝酸溶液至20mL。于消解液及试剂空白液中各加2mL柠檬酸铵溶液(200g/L),1mL盐酸羟胺溶液(200g/L)和2滴酚红指示液(1g/L),用氨水溶液(1+1)调至红色,再各加2mL氰化钾溶液(100g/L),混匀。各加5mL二硫腙使用液,剧烈振摇1min,静置分层后,三氯甲烷层经脱脂棉滤入1cm比色杯中,于波长510nm处测吸光度,与标准系列比较定量。
20、分析结果的表述
同13。
21、精密度
在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的10%。
22、其他
以称样量0.5g(或0.5mL)计算,方法的检出限为1mg/kg(或1mg/L),定量限为3mg/kg(或3mg/L)。


上一篇:GB/T 5009.17-2014食品中总汞及有机汞的测定
下一篇:无

扫描二维码
版权所有©广州格丹纳仪器有限公司 ![]() 粤公网安备 44011202000628号 粤ICP备14047970号
粤公网安备 44011202000628号 粤ICP备14047970号
分享到:







